CTF Dementia Research Fellowship Grant (2017-18)
Amantha Thathiah, PIND’s Dementia Researcher and University of Pittsburgh’s Assistant Professor of Neurobiology
In 2017, Clear Thoughts Foundation discovered one of Pittsburgh’s best kept secrets, The Pittsburgh Institute For Neurodegenerative Diseases (PIND). Located in the heart of Pittsburgh, the PIND takes a collaborative, interdisciplinary approach to the study of neurodegenerative diseases and their mechanisms, including Parkinson’s Disease, Amyotrophic Lateral Sclerosis (ALS), Huntington’s Disease, Stroke, and Dementia, such as Alzheimer’s Disease. Through basic, translation, and clinical research, the PIND seeks to ultimately apply basic laboratory findings in the clinic, transforming the latest scientific findings into new treatments and applications for those affected by neurodegenerative diseases.
The PIND’s dementia researcher and University of Pittsburgh’s Assistant Professor of Neurobiology, Amantha Thathiah has been the awarded grant recipient for both 2017 and 2018.

The Scientist Behind the Mission
Amantha has served as the Assistant Professor of Neurobiology at the University of Pittsburgh School of Medicine since 2016. She has been the recipient of the Cornelli Young Investigator Award for research in Alzheimer’s Disease and the Memory Loss in Alzheimer’s Disease (MEMOSAD) Award.
Amantha has produced many publications and is passionate about finding a cure for dementia such as Alzheimer’s disease, with her work focused on the Role of GPCRs in the AD brain.
“I think that a major challenge in AD is to gain a clearer understanding of the cellular processes that underlie disease pathogenesis and to link molecular and biochemical changes to specific clinical manifestations of disease in order to develop efficacious, mechanism-based therapeutic strategies. The project with CTF will address these challenges and provide fundamental salient insight into the involvement of GPCRs in disease progression.”
- Amantha Thathiah
Explore Thathiah’s Dementia Research
Alzheimer's disease is the most common type of dementia in people aged 65 or older and is one of the biggest medical and social challenges of our generation. Of the top ten leading causes of death worldwide, Alzheimer's disease is the only one that we cannot prevent, cure, or slow down.
In the United States alone, 5.3 million people have Alzheimer's disease, and ~480,000 new cases are diagnosed each year. Several approved drugs ameliorate some of the symptoms of Alzheimer's disease. Unfortunately, no current intervention strategy can modify the underlying disease mechanisms. Therefore, a major challenge for the future is to gain a clearer understanding of disease mechanisms in order to develop effective therapeutic strategies.
Alzheimer’s disease is clinically characterized by progressive brain cell loss and inflammation, memory impairment, cognitive deficits, and behavioral changes. Postmortem studies of the brains from patients reveal two pathological hallmarks of the disease: (1) amyloid plaques, composed of aggregates of the amyloid-beta protein, and (2) neurofibrillary tangles, composed of aggregates of the tau protein.
An imbalance between the production and clearance of amyloid-beta initiates a cascade of events, progressively leading to tau pathology, inflammation, brain cell loss, and, consequently, memory loss. A major challenge for the future is to gain a clearer understanding of disease mechanisms in order to develop effective therapeutic strategies.
Amyloid-β peptide (Aβ) is the principal protein component of the characteristic amyloid plaques in Alzheimer's disease patients. A compelling body of evidence compiled over more than 25 years points to Aβ as a key pathogenic feature in disease progression. The discovery that certain G protein–coupled receptors (GPCRs) can regulate Aβ production has offered fresh hope for Alzheimer’s disease drug discovery, as roughly half of current prescription drugs target GPCRs. We determine that the orphan GPCR GPR3 is involved the regulation of Aβ generation and amyloid plaque formation in different Alzheimer’s disease mouse models. Specifically, genetic deletion of the Gpr3 gene reduces the amyloid plaque burden and alleviates the cognitive deficits in mouse models of Alzheimer’s disease, indicating that GPR3 is involved in disease progression. These observations have potentially tremendous cell biological and therapeutic implications that could contribute fundamental insight into understanding GPCR dysfunction in Alzheimer’s disease. In collaboration with Clear Thoughts Foundation, we propose to determine GPCR profile in control and AD patient brain samples to establish the repertoire of aberrantly regulated GPCRs in AD. We will then investigate the functional relevance of changes in GPCR expression on amyloid pathology in in vitro and in vivo Alzheimer’s disease models. We anticipate that these studies will lead to novel insights into the role of GPCR in Alzheimer’s disease and identify putative novel therapeutic targets for Alzheimer’s disease.
During the most recent grant cycle (01/2018 – 12/2018), we had two specific research aims.
Specific Aim 1:
To determine whether the GPCRs that are differentially expressed in the human AD brain are involved in the proteolysis of APP.
Specific Aim 2:
To determine whether the identified GPCR(s) are involved in AD pathogenesis in the physiologically relevant context of human neurons.
We began cloning the validated GPCRs into the appropriate expression vectors, which allows us to investigate whether the identified GPCRs are indeed involved in the modulation of amyloid generation in cell culture models. Molecular cloning is not a precise science, and the cloning strategy for each of the identified GPCR requires a significant amount of time and optimization. Nevertheless, we were excited to determine whether any of the identified GPCRs are involved in amyloid generation, so we decided work in parallel, i.e. to test the GPCRs that were successfully cloned into appropriate expression vectors while cloning the remaining GPCRs. Although the GPCRs that we have tested thus far do not have robust effect on amyloid generation, the majority of the GPCRs identified in the RNA-Seq dataset have not been tested for their effect on amyloid generation. Collectively, in the last 2 years, we have identified and generated the toolbox to evaluate the GPCRs involved the pathophysiology of AD and which represent putative novel avenues for therapeutic intervention in AD. In looking towards the future, I have an excellent graduate student who is now working full-time on the CTF project.
Click to Enlarge
The first CTF grant (12/2016 – 11/2017) funded the RNA-Seq study, which identified GPCRs that are aberrantly expressed in the Alzheimer's Disease brain. Although preparing samples for an RNA-seq study does not take a tremendous amount time, analysis and validation of the huge dataset (~4 TB of data) that was generated does indeed require a tremendous amount of time. Parsing the data and identifying GPCRs targets was accomplished during the first grant cycle.
RNA - Seq is a deep sequencing-based method that allows the entire transcriptome to be surveyed in a very high- throughput and quantitative manner and has the power to accurately determine expression levels of weakly expressed genes. We have performed the RNA-Seq studies with control and AD patient samples. Based on the RNA-Seq studies, we determined that 19 GPCRs are differentially expressed in the human.
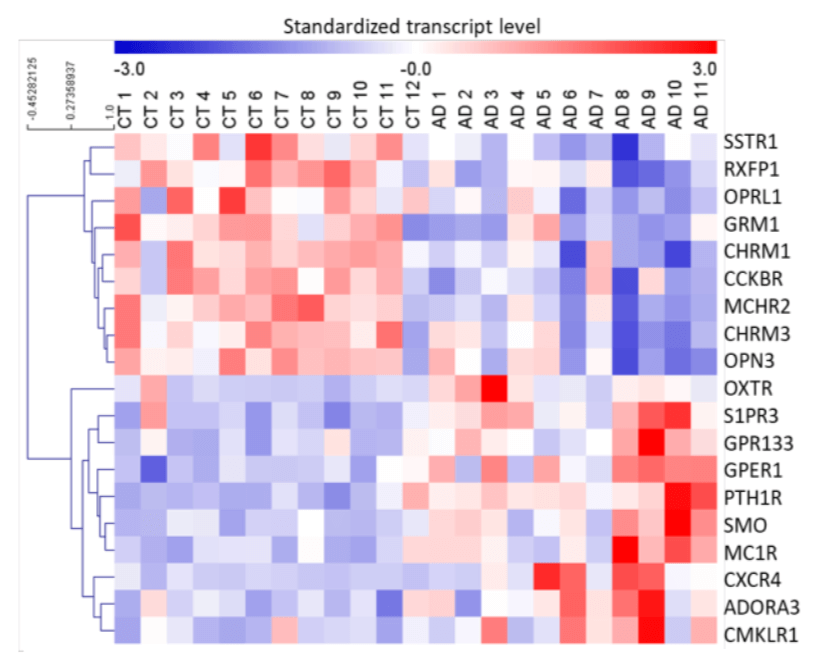 Figure 1. Differentially expressed GPCRs in brains of AD patients, revealed by RNA sequencing. Heat map of differentially expressed GPCRs in prefrontal cortex (PFC) of AD patients versus control (CT) subjects. P < 0.05 using Limma package with Benjamini-Hochberg correction for multiple testing. Standardized transcript levels are represented on a scale for which blue corresponds to lower levels of expression. The heat map was generated with MeV4.9.0.
Figure 1. Differentially expressed GPCRs in brains of AD patients, revealed by RNA sequencing. Heat map of differentially expressed GPCRs in prefrontal cortex (PFC) of AD patients versus control (CT) subjects. P < 0.05 using Limma package with Benjamini-Hochberg correction for multiple testing. Standardized transcript levels are represented on a scale for which blue corresponds to lower levels of expression. The heat map was generated with MeV4.9.0.
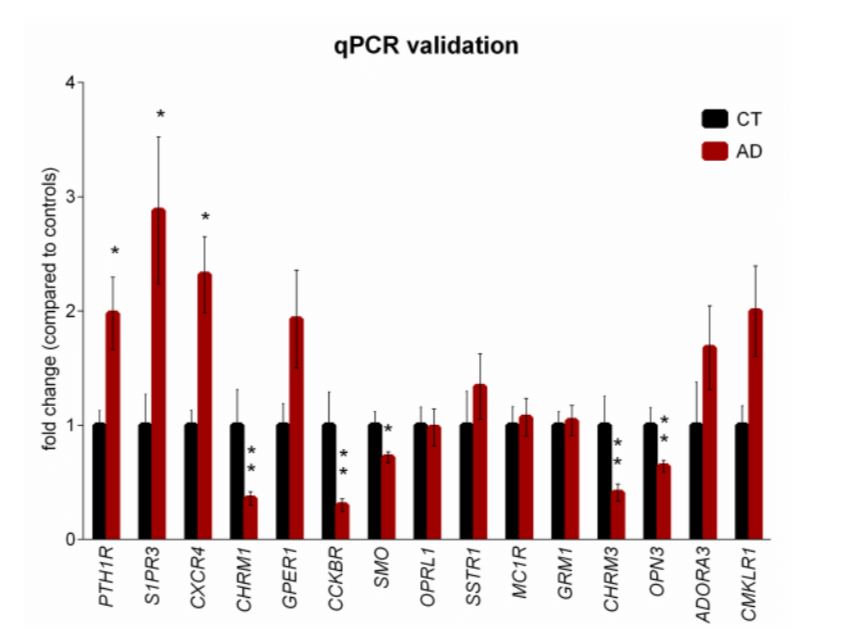 Figure 2. qPCR validation of selected differentially expressed GPCRs in brains of AD patients. GPCRs selected for validation by qPCR in brain samples from AD patients and CT subjects are represented as the fold change compared to the controls. The qPCR results were normalized to 2 internal controls, RPL5 and TAF7. *P < 0.05, **P < 0.01 by unpaired Student’s t test with Benjamini-Hochberg correction for multiple testing. n = 10 CT subjects, n = 24 AD patients. Error bars, s.e.m.
Figure 2. qPCR validation of selected differentially expressed GPCRs in brains of AD patients. GPCRs selected for validation by qPCR in brain samples from AD patients and CT subjects are represented as the fold change compared to the controls. The qPCR results were normalized to 2 internal controls, RPL5 and TAF7. *P < 0.05, **P < 0.01 by unpaired Student’s t test with Benjamini-Hochberg correction for multiple testing. n = 10 CT subjects, n = 24 AD patients. Error bars, s.e.m.
AD brain relative to control subjects (Fig. 1).
The level of sensitivity of the RNA-Seq approach is comparable to quantitative real-time polymerase chain reaction (qPCR) for its high sensitivity, specificity, and broad dynamic range. Therefore, we utilized qPCR to validate changes in GPCR expression observed by RNA-Seq on novel targets and previously identified GPCRs. The brain samples used for the RNA-Seq studies were also used for the validation studies, providing the most relevant basis for comparison. We designed two sets of primers for each differentially expressed gene. We then optimized the qPCR conditions for detection of each gene. To account for difference in the cellular neurodegenerative changes, we tested various internal control genes to establish genes with stable expression in control subjects and in AD patients. Expression levels of each gene was then normalized to each internal control. Based on the qPCR studies, we confirmed changes in expression of 15 GPCRs, (Fig. 2). We are confident that these GPCRs are differentially expressed in the AD brain relative to the control samples.
